ARTICLE in RSC Energy Advances : In-Depth Insight into Electrolytes
New Paper in Royal Society of Chemistry (RSC) Energy Advances
CLICK HERE TO ACCESS THE PAPER ; PDF Link

New Paper in Royal Society of Chemistry (RSC) Energy Advances
CLICK HERE TO ACCESS THE PAPER ; PDF Link
LINK To arXiv PREPRINT : CLICK HERE
New Paper by Dr. Vidushi Sharma :
Effects of Graphene Interface on Potassiation in a Graphene-Selenium Heterostructure Cathode for Potassium-Ion Batteries, ACS Applied Energy Materials, 2023

Selenium (Se) cathodes are an exciting emerging high energy density storage system for potassium-ion batteries (KIB), where potassiation reactions are less understood. Here, we present an atomic-level investigation of a KxSe cathode enclosed in hexagonal lattices of carbon (C) characteristic of a layered graphene matrix and multiwalled carbon nanotubes (MW-CNTs). Microstructural changes directed by the graphene−substrate in the KxSe cathode are contrasted with those in the graphene-free cathode. Graphene’s binding affinity for long-chain polyselenides (Se3 = −2.82 eV and Se2 = −2.646 eV) at low K concentrations and ability to induce enhanced reactivity between Se and K at high K concentrations are investigated. Furthermore, intercalation voltage for graphene-enclosed KxSe cathode reaction intermediates is calculated with K2Se as the final discharged product. Our results indicate a single-step reaction near a voltage of 1.55 V between K and Se cathode. Findings in the paper suggest that operating at higher voltages (∼2 V) could result in the formation of reaction intermediates where intercalation/deintercalation of K could be a challenge, and therefore cause irreversible capacity losses in the battery. The primary issue here is the modulating favorability of graphene surface toward discharging of Se cathode due to its differential preferences for K−Se reaction intermediates. A comparison with a graphene-free cathode highlights the substantial changes a van der Waals (vdW) graphene interface can bring in the atomic structure and electrochemistry of the KxSe cathode.
Among intercalation, alloying, and conversion battery chemistries, the intercalation chemistry is most widely used in commercial applications due to its superior reversibility, round trip efficiency, and stability, albeit at the expense of reduced specific capacity. While intercalation hosts for monovalent ions (e.g., lithium and sodium) are well developed, the jury is still out on the best available intercalation host materials for multivalent ions such as magnesium, zinc, calcium, and aluminum. In multivalent systems, it is challenging to find electrode materials that can act as a durable host, and accommodate large number of ions, while also permitting fast diffusion kinetics. In this perspective, the electrochemical performance of five distinct class of materials (prussian blue analogues, sodium super ionic conductors organic, layered, and open-tunnel oxides) for multivalent ion storage is evaluated. The analysis reveals that open-tunnel oxides show noticeably superior performance in multivalent ion batteries. Herein, the underlying reasons for this are discussed and the case is made for an in-depth machine-learning-driven “materials exploration effort” directed toward discovery of new open-tunneled oxides that could lead to vastly superior multivalent ion batteries.
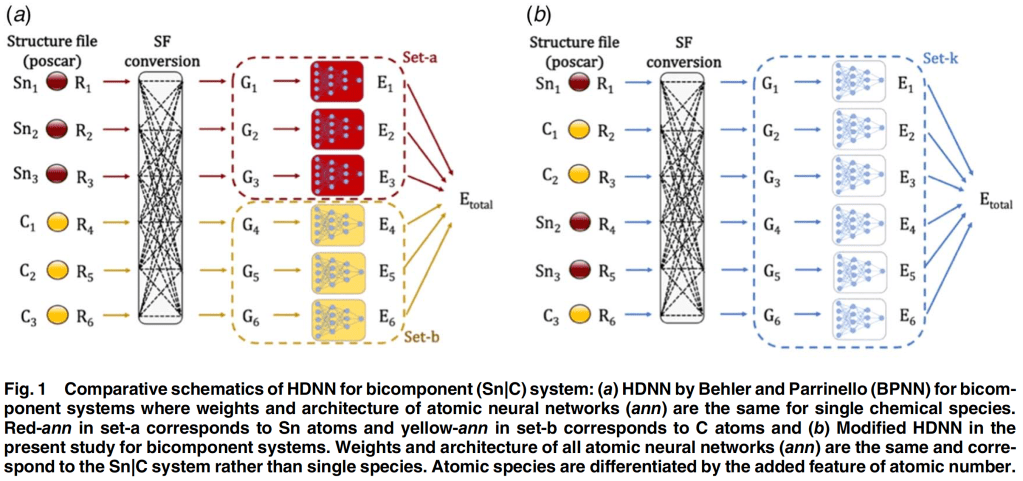
We recently published this article in ASME Special Issue on “Emerging Investigators in Electrochemical Energy Conversion and Storage 2022” :
V. Sharma, D. Datta, Developing Potential Energy Surfaces for Graphene-Based 2D–3D Interfaces From Modified High-Dimensional Neural Networks for Applications in Energy Storage, ASME Journal of Electrochemical Energy Conversion and Storage, 19(4):041006, 2022
Abstract : Designing a new heterostructure electrode has many challenges associated with interface engineering. Demanding simulation resources and lack of heterostructure databases continue to be a barrier to understanding the chemistry and mechanics of complex interfaces using simulations. Mixed-dimensional heterostructures composed of two-dimensional (2D) and three-dimensional (3D) materials are undisputed next-generation materials for engineered devices due to their changeable properties. The present work computationally investigates the interface between 2D graphene and 3D tin (Sn) systems with density functional theory (DFT) method. This computationally demanding simulation data is further used to develop machine learning (ML)-based potential energy surfaces (PES). The approach to developing PES for complex interface systems in the light of limited data and the transferability of such models has been discussed. To develop PES for graphene-tin interface systems, high-dimensional neural networks (HDNN) are used that rely on atom-centered symmetry function to represent structural information. HDNN are modified to train on the total energies of the interface system rather than atomic energies. The performance of modified HDNN trained on 5789 interface structures of graphene|Sn is tested on new interfaces of the same material pair with varying levels of structural deviations from the training dataset. Root-mean-squared error (RMSE) for test interfaces fall in the range of 0.01–0.45 eV/atom, depending on the structural deviations from the reference training dataset. By avoiding incorrect decomposition of total energy into atomic energies, modified HDNN model is shown to obtain higher accuracy and transferability despite a limited dataset. Improved accuracy in the ML-based modeling approach promises cost-effective means of designing interfaces in heterostructure energy storage systems with higher cycle life and stability.