SDSC Media Coverage : Graphene, Tin Combo Shows Promise
San Diego Supercomputer Center (SDSC) highlighted our work: Graphene, Tin Combo Shows Promise for Solar Panels, Artificial Muscles and More
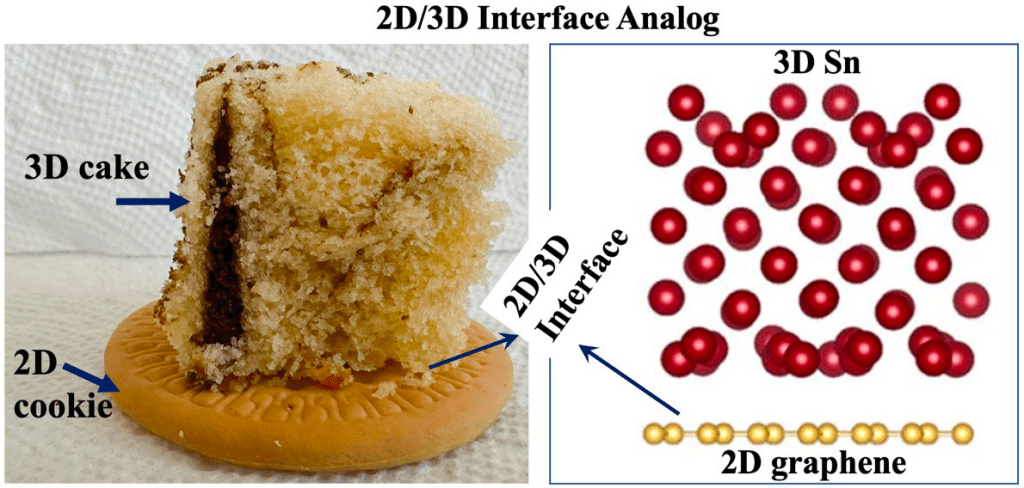
Check the related paper : Developing Potential Energy Surfaces for Graphene-Based 2D–3D Interfaces From Modified High-Dimensional Neural Networks for Applications in Energy Storage, ASME Journal of Electrochemical Energy Conversion and Storage, 19(4):041006, 2022
