First Paper of 2024 : JOM – TMS
Datta, D. Electro-Chemo-Mechanical Modeling of Multiscale Active Materials for Next-Generation Energy Storage: Opportunities and Challenges. JOM (2024).
LINK : https://doi.org/10.1007/s11837-023-06335-y
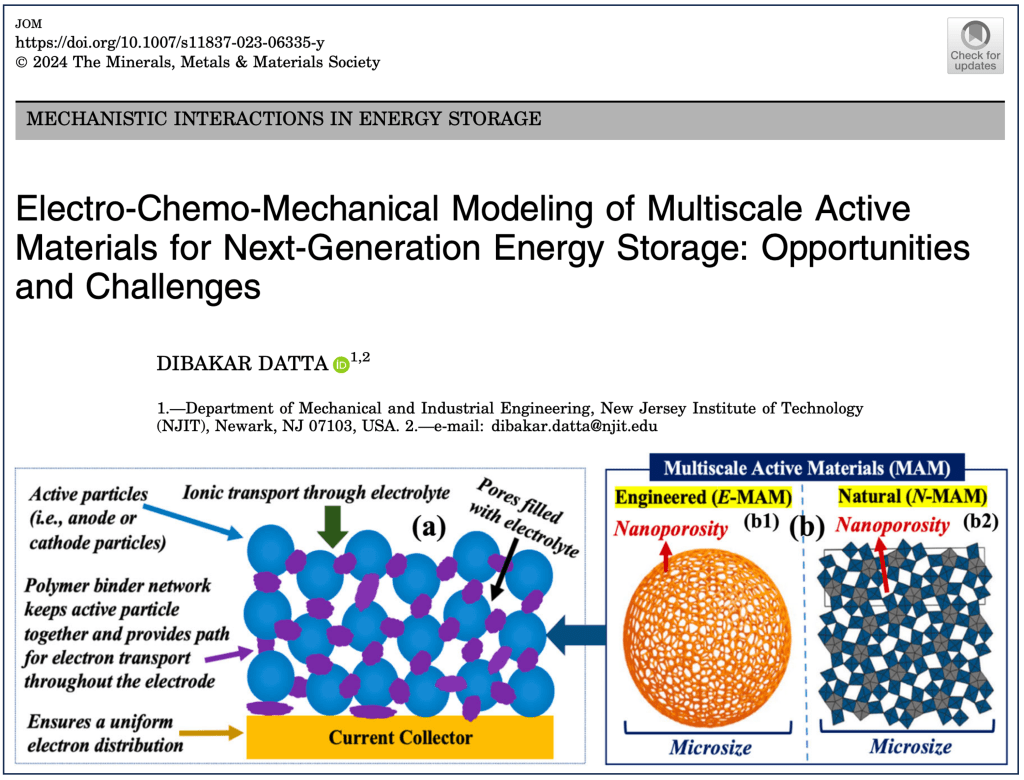
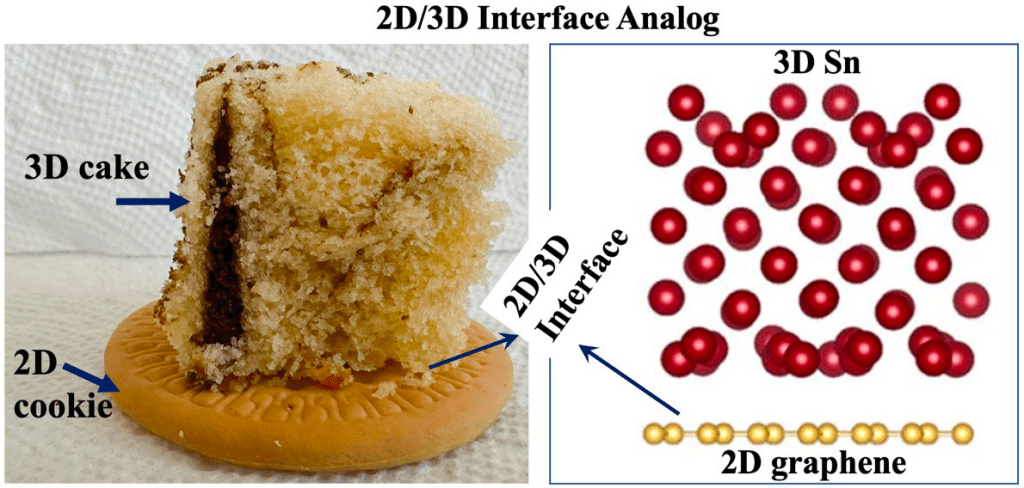
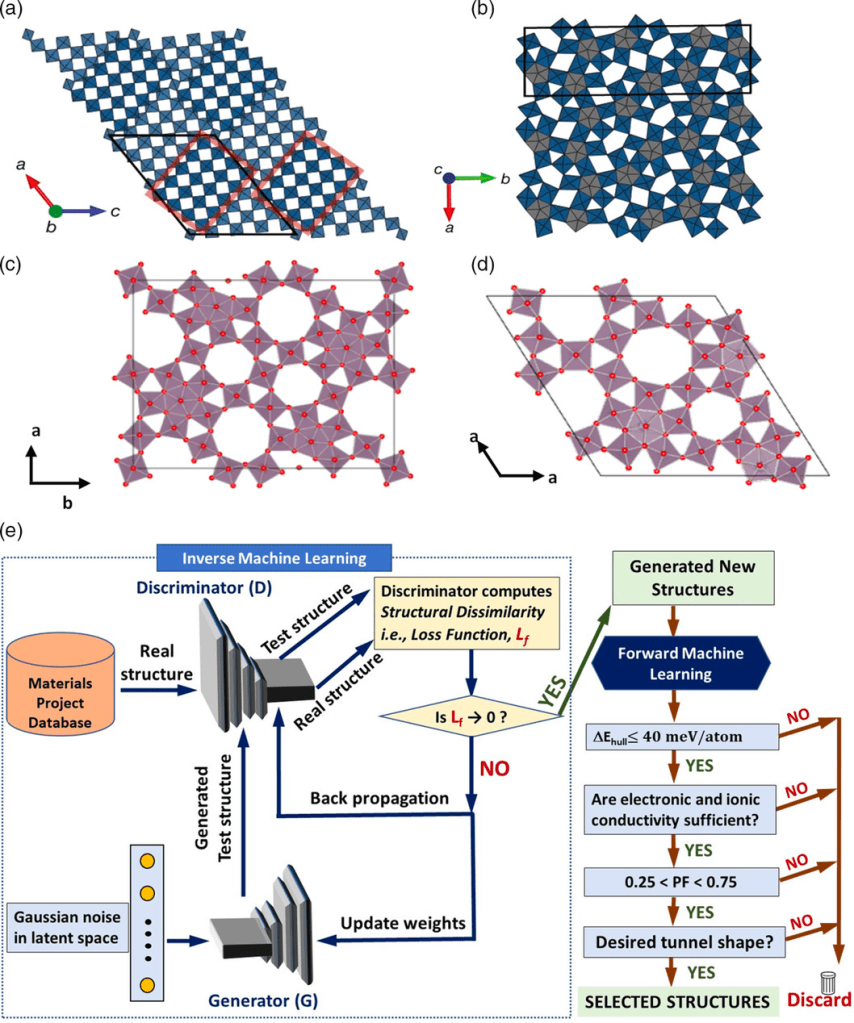
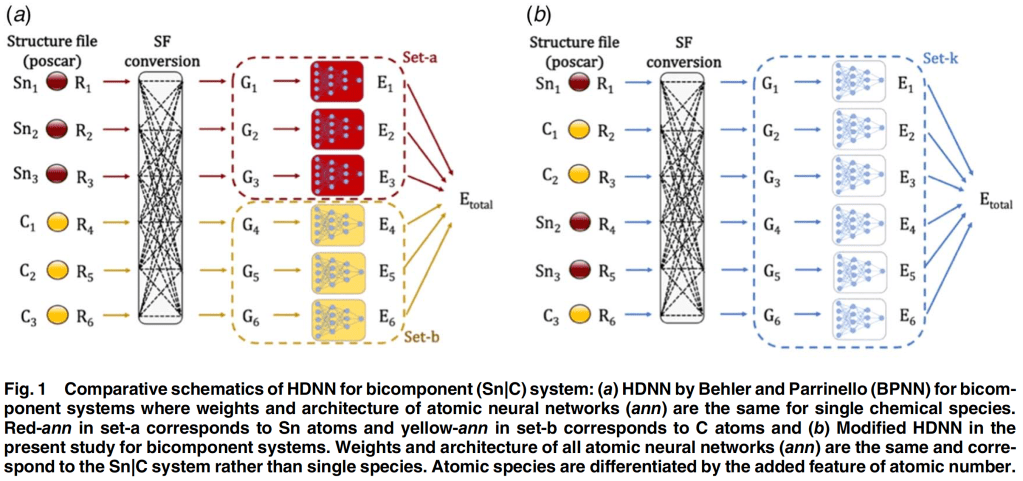
Although lithium-ion batteries represent the best available rechargeable battery technology, a significant energy and power density gap exists between LIBs and petrol/gasoline. The battery electrodes comprise a mixture of active materials particles, conductive carbon, and binder additives deposited onto a current collector. Although this basic design has persisted for decades, the active material particle’s desired size scale is debated. Traditionally, microparticles have been used in batteries. Advances in nanotechnology have spurred interest in deploying nanoparticles as active materials. However, despite many efforts in nano, industries still primarily use ‘old’ microparticles. Most importantly, the battery industry is unlikely to replace microstructures with nanometer-sized analogs. This poses an important question: Is there a place for nanostructure in battery design due to irreplaceable microstructure? The way forward lies in multiscale active materials, microscale structures with built-in nanoscale features, such as microparticles assembled from nanoscale building blocks or patterned with engineered or natural nanopores. Although experimental strides have been made in developing such materials, computational progress in this domain remains limited and, in some cases, negligible. However, the fields hold immense computational potential, presenting a multitude of opportunities. This perspective highlights the existing gaps in modeling multiscale active materials and delineates various open challenges in the realm of electro-chemo-mechanical modeling. By doing so, it aims to inspire computational research within this field and promote synergistic collaborative efforts between computational and experimental researchers.